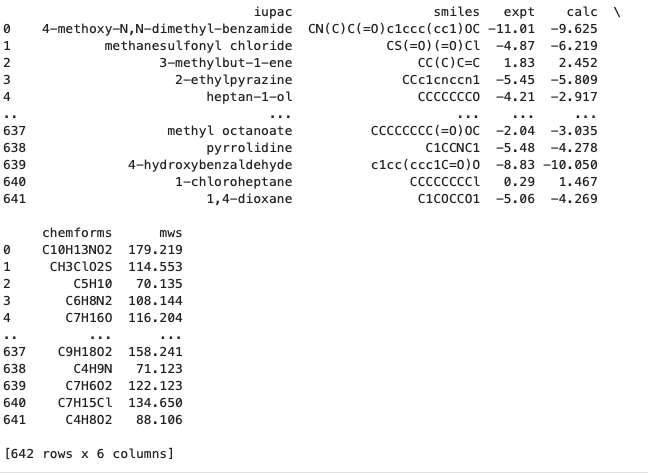
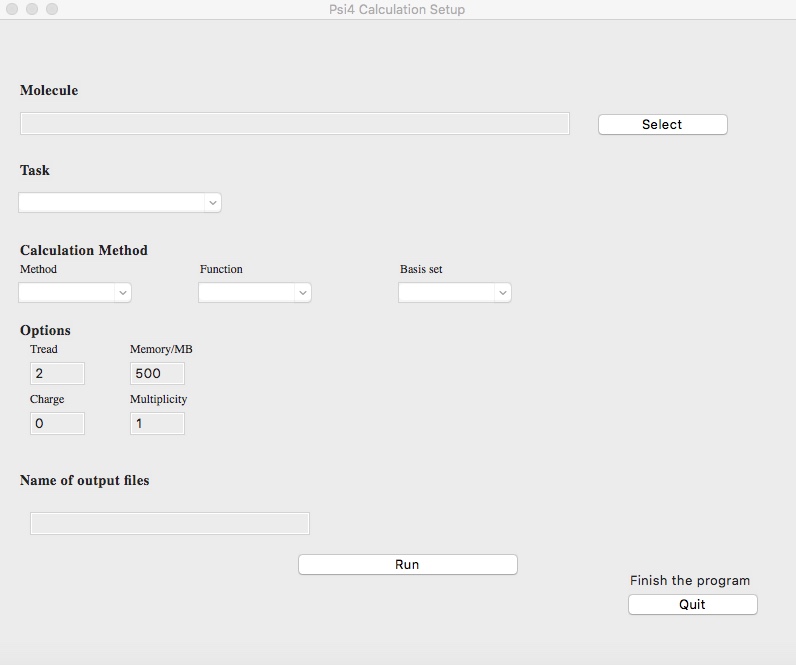
前回に引き続きCustumTkinterを利用した軌道情報の自動計算アプリの紹介です。
今回は量子化学計算用の処理関数の設定コードの紹介です。構造最適化、振動計算、分子軌道の処理関数を作成しています。
まずは構造最適化(Geom_Opt)です。CustumTkinter(コードは後の記事で紹介予定)での入力値から計算方法、基底関数などデータを読み取っています。計算手法はHF、DFT、MP2を選択できるようにしていますので、それぞれの入力値に合わせて、最適化計算できるようにif文で分岐させています。今回は自動的に多くのデータをある程度の精度で処理する必要があるので、構造最適化の最大回数(geom_maxiter)、収束しきい値(optking__g_convergence)をオプションで敢えて緩めに設定しています。
続いて振動計算(Vib_Calc)です。構造最適化と同じく入力値から計算手法を選択して実施できるようにしています。計算結果はエネルギー(energy)と波動関数情報(wfn)に格納した上で、振動数情報をwfnから振動数情報を取り出しています(振動数は直接アウトプットされないため)。振動数のリスト(freqlist)から有用な振動数を判定で取り出していますが(freqposi, freqnega)、今回は虚数解がないかどうか(安定構造か遷移状態か)を重視して、あとで使えるよう負の振動数(freqnega)をglobal指定しています。
最後は分子軌道(MO_anal)です。分子軌道の計算(エネルギー計算)も入力値から選択して実施できるようになっています(ただし、構造最適化、振動計算、エネルギー計算は入力値ですべて同じ手法で実施されます)。また表示させる軌道エネルギーの数も入力値(orb)に応じて変えられるようになっています。軌道エネルギーも直接表示されるわけではありませんので、波動関数情報(wfn)から適宜取り出して、HOMO、LUMOのリストとして格納しています。
def Geom_Opt(): # Geometry optimization # getting meth(calculation method), func(function), base(basis set) meth=method_sv.get() func=function_sv.get() base=baseset_sv.get() psi4.set_options({'geom_maxiter': 50, 'optking__g_convergence': 'gau_loose',}) # geometry optimize calculation (method in HF and MP2, function in DFT) if meth=='HF': psi4.optimize(meth+'/'+base, molecule=molgeom) elif meth=='DFT': psi4.optimize(func+'/'+base, molecule=molgeom) elif meth=='MP2': psi4.optimize(meth+'/'+base, molecule=molgeom) optimized_geom = molgeom.save_string_xyz_file() def Vib_Calc(): # Frequency calculation meth=method_sv.get() func=function_sv.get() base=baseset_sv.get() # Frequency calculation (method in HF and MP2, function in DFT) if meth=='HF': energy, wfn = psi4.frequency(meth+'/'+base, molecule=molgeom, return_wfn=True) elif meth=='DFT': energy, wfn = psi4.frequency(func+'/'+base, molecule=molgeom, return_wfn=True) elif meth=='MP2': energy, wfn = psi4.frequency(meth+'/'+base, molecule=molgeom, return_wfn=True) # making frequency list freqlist = np.array(wfn.frequencies()) freqposi=freqlist[(freqlist>1)&(freqlist<5000)] global freqnega freqnega=freqlist[(freqlist<0)] return freqposi, freqnega def MO_anal(): # Molecular orbital analysis meth=method_sv.get() func=function_sv.get() base=baseset_sv.get() orb=orb_input.get() global wfn global HOMOdata, LUMOdata global Hindex, Lindex # Molecular orbital analysis (method in HF and MP2, function in DFT) if meth=='HF': energy, wfn = psi4.energy(meth+'/'+base, molecule=molgeom, return_wfn=True) elif meth=='DFT': energy, wfn = psi4.energy(func+'/'+base, molecule=molgeom, return_wfn=True) elif meth=='MP2': energy, wfn = psi4.energy(meth+'/'+base, molecule=molgeom, return_wfn=True) # orbital_level : number of display level orbital_level=orb orblev=np.array(list(range(((orbital_level)+1)))) HOMOdata=[] LUMOdata=[] Hindex=[] Lindex=[] LUMO_idx=wfn.nalpha() HOMO_idx=LUMO_idx-1 for i in orblev: nexthomo=HOMO_idx-(i) nextlumo=LUMO_idx+(i) nhomolev=wfn.epsilon_a_subset("AO","ALL").np[nexthomo] nlumolev=wfn.epsilon_a_subset("AO","ALL").np[nextlumo] HOMOdata.append(nhomolev) LUMOdata.append(nlumolev) Hindex.append('homo-'+str(i)) Lindex.append('lumo+'+str(i)) return HOMOdata, LUMOdata, Hindex, Lindex
以上、今回は量子化学計算用の処理関数について紹介しました。次回は主計算の処理関数について紹介したいと思います。